Синдром (или болезнь) Марфана – это редкое генетическое заболевание, наследующееся по аутосомно-доминантному типу и обусловленное структурными дефектами соединительной ткани, которые приводят к поражению преимущественно опорно-двигательной, зрительной и сердечно-сосудистой систем организма. Частота встречаемости такой наследственной коллагенопатии невысока и по разным данным статистики составляет не более 1 случая на 10 или 20 тысяч человек. Заболевание может выявляться у людей любого пола и не имеет характерной расовой определенности.
В этой статье вы сможете ознакомиться с причинами, разновидностями, проявлениями, способами диагностики и лечения, прогнозами синдрома Марфана. Эта информация поможет составить представление об этом наследственном заболевании, и вы сможете задать возникающие вопросы лечащему врачу.
Клинические проявления этого синдрома весьма полиморфны. Люди с болезнью Марфана могут иметь высокий рост, диспропорционально длинные конечности, удлиненные и тонкие (как их называют «паучьи») пальцы, астеническое телосложение, арковидное небо, удлиненный череп, скученные зубы и глубоко посаженные глаза. Нередко у них обнаруживается деформированная грудная клетка, кифосколиоз, маленькие размеры челюсти, эктопия хрусталика, близорукость, плоскостопие, аневризмы аорты и протрузия вертлужной впадины.
Такой широкий фенотипический набор проявлений может быть выражен как в легких и почти неотличимых от состояния нормы, так и в тяжелых, выраженных и быстропрогрессирующих формах. Подобная вариабельность объясняется крайним разнообразием мутаций в гене FBN1 и некоторых других генах (TGFBR-2 и др.).
Отмечается, что в 75 % случаев синдром Марфана передается по семейному типу наследования и только у 15 % детей мутация проявляется впервые. Кроме этого, специалисты выделяют тот факт, что риск рождения ребенка с таким заболеванием увеличивается в тех парах, в которых возраст отца более 35 лет.
Немного истории и интересных фактов

Впервые заболевание было описано в 1886 году педиатром из Франции Антуаном Марфаном. Именно им был рассмотрен клинический случай девочки, имеющей «паучьи» пальцы и тонкие длинные ноги. Такие аномалии скелета быстро прогрессировали. Впоследствии заболевание было названо его именем. Ген, вызывающий развитие признаков синдрома, был впервые выявлен только в 1991 году Франческо Рамиресом в центре Маунт Синай (США, Нью-Йорк).
Наблюдения показывают, что этот недуг нередко выявлялся у знаменитых людей. Великий и всемирно известный скрипач Никколо Паганини, знаменитый сказочник Ханс Христиан Андерсен, президент США Авраам Линкольн, композитор Сергей Рахманинов, вокалист Ramones Джоуи Рамон и певец Трой Сиван, актер Винсент Скьявелли, первая модель Лесли Хорнби, фронтмэны групп Deerhunter и Of Mice & Men Брэдфорд Кокс и Остин Карлайл – это далеко не все популярные люди с таким недугом.
Предполагается, что такие неординарные способности могут вызываться характерным для синдрома Марфана высоким выбросом адреналина, который провоцирует гиперактивность и в некоторых случаях может вызывать развитие талантов.
Причины
Это генетическое заболевание преимущественно наследуется по доминантному типу и вызывается передачей ребенку аномального гена FBN1, отвечающего за синтез фибрилина-1. Этот протеин отвечает за эластичность и сократимость соединительной ткани, а его дефицит вызывает утрату ее прочности и упругости. Из-за этого соединительная ткань не может выдерживать возлагающиеся на нее физиологические нагрузки. Таким изменениям в большей степени подвержены стенки кровеносных сосудов и связочный аппарат (в первую очередь поражается аорта и цинновая связка глаза).
Протеин фибрилин-1 выполняет не только опорные функции для тканей, но и связывается с другим белком. Такая комбинация формирует фактор роста TGF- β, оказывающий негативное воздействие на тонус мышц сосудов. Он накапливается в избыточном количестве в тканях легких, клапанах сердца и аорте. Впоследствии их ткани ослабляются и у больного возникают характерные для заболевания проявления.
Классификация
Специалисты выделяют две основные формы этого синдрома:
- стертая – у больного наблюдаются слабовыраженные поражения 1 или 2 систем;
- выраженная – слабовыраженному поражению подвергаются 3 системы или выраженные изменения выявляются в 1 системе, или выраженные поражения присутствуют в 2-3 и более системах.
Поражения при синдроме Марфана могут быть выражены в легкой, средней или тяжелой формах. Течение заболевания может быть стабильным или прогрессирующим.
Симптомы
Наиболее характерным признаком синдрома Марфана является сочетание поражений опорно-двигательной, зрительной и сердечно-сосудистой систем. Сроки их появления вариабельны, а проявления многообразны.
Обычно для постановки диагноза бывает достаточно присутствия следующих признаков:
- непропорционально длинные конечности;
- аневризма аорты;
- одно- или двухсторонняя эктопия хрусталика.
Однако кроме этих признаков существует еще около 30 других проявлений синдрома.
Поражения скелета
У людей с синдромом Марфана могут выявляться следующие изменения в опорно-двигательной системе:
- рост намного выше среднего;
- астенический тип телосложения;
- длинный и узкий лицевой скелет;
- длинные конечности и пальцы;
- искривления позвоночного столба (спондилолистез, сколиоз, кифоз и др.);
- воронкообразная или килевидная деформация грудной клетки;
- маленький размер челюсти;
- аркоподобное высокое небо;
- чрезмерная гибкость и подвижность суставов;
- молоткообразная деформация пальцев стоп;
- протрузия вертлужной впадины;
- плоскостопие;
- патологии прикуса.
Средний рост людей с таким заболеванием при рождении может составлять у девочек 52,5 см (во взрослом возрасте около 175 см), у мальчиков 53 см (во взрослом возрасте около 191 см).
Из-за высокого неба и малых размеров челюсти у людей с синдромом Марфана могут возникать нарушения речи. Поражения скелета и суставных структур приводят к появлению артралгий и миалгий. Позднее такие изменения повышают риск развития раннего остеоартрита.
Поражения органов зрения
Одно- или двухсторонняя эктопия хрусталика при синдроме Марфана выявляется у 80 % пациентов. Обычно у таких людей развивается близорукость и астигматизм, но в некоторых случаях возникает и дальнозоркость. Чаще нарушения зрения происходят на 4-м году жизни ребенка. Далее они устойчиво прогрессируют.
Кроме этих заболеваний, при синдроме Марфана могут выявляться следующие патологии органов зрения:
- косоглазие;
- гипоплазия цилиарной мышцы;
- колобома радужной оболочки;
- увеличение размера и уплощение роговицы;
- изменение диаметра сосудов сетчатки;
- формирование катаракты;
- возникновение глаукомы в молодом возрасте.
Поражения сердца и сосудов
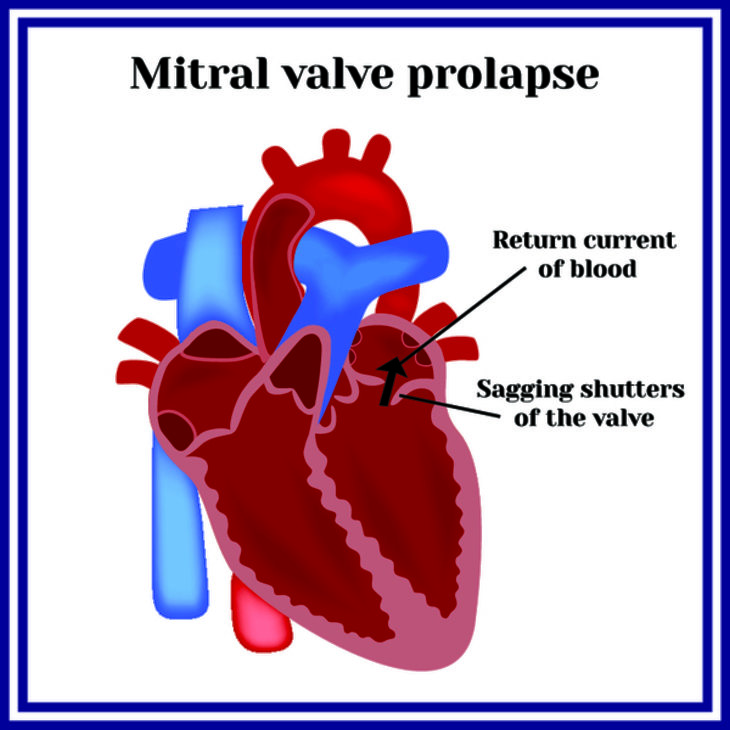
Доминирующими и наиболее опасными проявлениями синдрома Марфана становятся признаки поражений сердца и сосудов. Появляющиеся из-за повреждения структуры стенок сосудов эластического типа изменения (особенно аорты и легочной артерии) и пороки развития клапанов, перегородок сердца вызывают следующие симптомы:
- быстрое наступление усталости;
- учащенное сердцебиение;
- стенокардические боли с локализацией в спине, верхней конечности или плече;
- холодные руки и ноги;
- аритмии;
- одышка.
При выслушивании тонов сердца у таких пациентов могут определяться шумы, а при выполнении ЭКГ выявляются признаки стенокардии. При синдроме Марфана может развиваться кистозная медиальная дегенерация митрального или аортального клапана, приводящая к пролапсу этих клапанных структур. Кроме этого, у плода, унаследовавшего мутированные гены, с высокой долей вероятности могут развиваться врожденные пороки сердца. Иногда, при неблагоприятном течении такой неонатальной формы синдрома, у ребенка возникает прогрессирующая сердечная недостаточность, приводящая к летальному исходу до года жизни.
Однако наиболее характерным для синдрома Марфана поражением сердечно-сосудистой системы обычно становится прогрессирующее расширение, расслоение всходящей части аорты и появление на ней аневризм. Такие изменения провоцируются ослаблением соединительной ткани, приводящим к кистозной дегенерации сосудистой стенки. Быстрое прогрессирование таких поражений аорты может охватывать всю ее длину и ответвляющиеся от нее сосуды. Нередко такое осложненное течение патологии приводит к смертельному исходу.
Поражения центральной нервной системы
Одним из последствий синдрома Марфана может становиться дуральная эктазия, вызывающаяся растяжением и вытяжением соединительной ткани (оболочки), обволакивающей спинной мозг. Впоследствии эта патология может приводить к появлению болей и дискомфортных ощущений в брюшной полости или к слабости и неподвижности нижних конечностей.
Интеллектуальное развитие и состояние психики
У большинства детей уровень интеллекта соответствует норме и IQ составляет 85-115 единиц. У некоторых лиц с таким наследственным заболеванием уровень IQ существенно превышает верхние границы нормы.
Иногда у людей с синдромом Марфана могут присутствовать признаки неравномерной интеллектуальной деятельности и некоторые особенности личности, выражающиеся в завышенной самооценке, чрезмерной эмоциональности, плаксивости и раздражительности.
Поражения легких
Больные с синдромом Марфана не всегда имеют проблемы с легкими. Однако в некоторых случаях слабость соединительной ткани альвеол приводит к их удлинению и перерастяжению. Впоследствии у таких больных может возникать спонтанный пневмоторакс, эмфизема легких и дыхательная недостаточность. При отсутствии своевременной помощи и лечения такие патологии могут становиться причиной наступления летального исхода.
Кроме этого, у людей с синдромом Марфана может наблюдаться ночное апноэ, сопровождающееся прекращением дыхания во сне на 10 секунд и более.
Поражения других систем
Кроме вышеописанных проявлений синдрома Марфана в некоторых случаях могут выявляться следующие изменения в других органах и системах:
- атрофические стрии на коже;
- часто рецидивирующие бедренные и паховые грыжи;
- предрасположенность к растяжению и разрывам связок, подвывихам и вывихам;
- аномальное расположение почек;
- варикозное расширение сосудов;
- опущение матки и мочевого пузыря.
Общее состояние
Большинство детей с синдромом Марфана тяжело переносят физическую нагрузку и после нее нередко ощущают боли в мышцах. Мышцы у таких больных могут быть недоразвитыми.
На фоне психоэмоционального перенапряжения у детей могут периодически возникать приступы мигренеподобной головной боли. Кроме этого, нередко ощущается слабость и признаки гипотонии.
Диагностика
Для выявления синдрома Марфана врач проводит тщательное изучение семейного анамнеза, физикальный осмотр больного и назначает ЭКГ, Эхо-КГ, молекулярно-генетический анализ, рентгенографическое и офтальмологическое обследование.
Основными диагностическими признаками синдрома Марфана являются следующие отклонения:
- расширение и/или расслоение восходящего отдела аорты;
- эктазия твердой мозговой оболочки;
- требующая лечения деформация грудной клетки;
- эктопия хрусталика;
- сколиоз (или спондилолистез);
- соотношение верхнего сегмента тела к нижнему менее 0,86 или размаха верхних конечностей к росту более 1,05;
- плоскостопие;
- протрузия вертлужной впадины.
Случаи семейного анамнеза являются дополнительными признаками заболевания, а остальные проявления синдрома Марфана относятся к малым диагностическим критериям.
Для окончательной постановки диагноза необходимо выявление по одному главному критерию в двух системах организма и одного малого в третьей системе или наличие 4-х главных критериев в скелете.
Кроме этого, выполняются тесты для определения характерных фенотипических признаков:
- соотношение длины кисти и роста (более 11 %);
- длина среднего пальца (более 10 см);
- индекс Варге и др.
Для выявления патологий сердца и сосудов проводят:
- ЭКГ – определяются признаки стенокардии, аритмии, выраженная гипертрофия миокарда;
- Эхо-КГ – выявляется расширение аорты, увеличение левого желудочка, пролапс митрального клапана, вызванная пролапсом клапана регургитация, разрывы хорд, аневризмы аорты.
При подозрениях на расслоение и аневризму аорты рекомендуется проведение аортографии.
Для исследования нарушений со стороны зрения проводятся следующие исследования:
- офтальмоскопия;
- биомикроскопия.
Изменения в скелете выявляются при помощи рентгенографии. Для определения эктазии твердой мозговой оболочки выполняется МРТ позвоночного столба.
При синдроме Марфана лабораторные исследования позволяют выявить увеличение почечной экскреции глюкозоаминогликанов и их фракций (метаболитов соединительной ткани) в 2 раза и более. При помощи метода прямого автоматического секвенирования ДНК определяются мутации в гене FBN1.
Дифференциальная диагностика синдрома Марфана проводится со следующими заболеваниями:
- синдром Билса;
- гомоцистонурия;
- синдром Шпринтцена – Гольдберга;
- синдром Элерса-Данлоса;
- синдром Стиклера;
- МАSS фенотип;
- синдром Лойса-Дитца;
- множественная эндокринная неоплазия типа 2В (МЕН II).
Лечение
Пока ученые не смогли найти средство для устранения нарушений развития соединительной ткани у больных с синдромом Марфана. Именно поэтому на сегодня основная цель терапии этого генетического заболевания направлена на профилактику прогрессирования недуга и возникновения его осложнений.
Медикаментозная терапия
При выявлении расширения аорты до 4 см или признаков пролапса митрального клапана больному назначаются β-адреноблокаторы, способствующие уменьшению нагрузки на этот крупный сосуд. Как правило, чаще рекомендуется прием этих средств длительного действия. Дозировка препарата назначается индивидуально (например, доза пропранолола может составлять от 40 до 200 мг/день, а атенолола – 25-150 мг). Кроме β-адреноблокаторов, для стабилизации работы сердца и сосудов могут дополнительно назначаться антагонисты кальция или ингибиторы АПФ.
При значительных дефектах скелета в организме наблюдается дефицит некоторых белков и микроэлементов, участвующих в строении соединительной ткани. Для восполнения их недостатка пациентам с синдромом Марфана могут назначаться препараты и БАДы на основе магния, цинка, кальция и меди, колекальциферола и гиалуроновой кислоты.
При выявлении в крови больного повышенного уровня соматотропного гормона, провоцирующего избыточный рост, в питание детей рекомендуется включать высокожировые энпиты класса Омега-3. Их введение в рацион позволяет несколько тормозить секрецию соматотропина и замедлять темп роста.
Девочкам с таким недугом при стремительном росте иногда может назначаться прием средств на основе прогестерона и эстрогена. Их обычно начинают применять в 10-летнем возрасте (для ускорения наступления полового созревания и более быстрого прекращения роста).
Кроме этого, пациентам (детям и взрослым) с синдромом Марфана могут назначаться следующие благотворно влияющие на соединительную ткань средства:
- аскорбиновая кислота;
- Глюкозамин сульфат;
- Хондроитин сульфат;
- Карнитина хлорид;
- L-лизин;
- токоферол;
- витаминно-минеральные комплексы.
Такие препараты принимаются курсами, и план их проведения составляется для каждого пациента индивидуально.
В некоторых случаях при синдроме Марфана требуется проведение стоматологических манипуляций и хирургических операций. Таким пациентам для профилактики возможных тромбозов и инфекционного эндокардита в послеоперационном периоде назначаются антикоагулянты и антибиотики.
Для коррекции нарушений зрения пациентам с синдромом Марфана рекомендуется подбор контактных линз или очков. При необходимости для лечения могут проводиться различные офтальмологические операции.
Изменение образа жизни

Пациентам с синдромом Марфана рекомендуется:
- некоторое ограничение физических нагрузок – они должны быть низкого или среднего уровня;
- отказ от некоторых видов физической активности (спортивные игры, требующие резких толчков или бросательных движений, подводное плавание, велосипедные гонки, спринтерские забеги и т. п.);
- решение о занятиях спортом всегда принимать совместно с лечащим врачом;
- частое обследование у кардиолога, окулиста, ортопеда и терапевта;
- при планировании зачатия ребенка провести консультацию у генетика;
- женщинам, планирующим беременность, следует быть готовыми к проведению досрочного родоразрешения при помощи кесарева сечения.
Хирургическое лечение
Для устранения поражений сердца и сосудов при синдроме Марфана могут проводиться следующие кардиохирургические вмешательства:
- пластика части аорты эндо- или экзопротезом (при ее расширении до более 6 см, расслоении и аневризмах);
- имплантация митрального клапана (проводится только при быстром прогрессировании регургитации до выраженной или развитии левожелудочковой недостаточности).
По данным статистики внедрение в практику кардиохирургов современных методик позволяет снижать риск послеоперационной смертности больных с синдромом Марфана, после их выполнения они становятся трудоспособными. Кроме этого, своевременное проведение таких вмешательств позволяет продлевать продолжительность жизни таких пациентов до 60-70 лет.
При необходимости для устранения нарушений со стороны зрения проводятся офтальмологические операции:
- лазерная коррекция миопии;
- хирургическое или лазерное устранение катаракты или глаукомы;
- имплантация искусственного хрусталика.
Вопрос о необходимости коррекций деформаций скелета решается консилиумом врачей. Такие вмешательства являются травматичными и нередко сопровождаются развитием тяжелых послеоперационных осложнений (перикардитов, плевритов, пневмоний). Вопрос о целесообразности их проведения неоднократно обсуждался на многочисленных симпозиумах, посвященных коллагенопатиям. Многие специалисты придерживаются мнения, отрицающего необходимость подобного хирургического лечения. Однако в тяжелых случаях такие корректирующие операции по стабилизации позвоночного столба, торакопластике или эндопротезированию тазобедренных суставов могут выполняться.
Прогноз
Продолжительность жизни людей с этим синдромом во многом определяется тяжестью поражений сердца и сосудов. Высокий риск наступления внезапной смерти и уменьшение продолжительности жизни до 40-50 лет наблюдается среди тех пациентов, которым не было назначено своевременное медикаментозное или хирургическое лечение. Вовремя проведенные кардиохирургические операции позволяют продлить жизнь больного до 60-70 лет, улучшают качество жизни и возвращают трудоспособность.
К какому врачу обратиться
При подозрении на наличие синдрома Марфана следует обратиться к врачу-генетику. После проведения диагностики к наблюдению и лечению пациента подключаются и другие специалисты: кардиолог, ортопед, офтальмолог и терапевт. При необходимости проведения корригирующих операций на сердце и сосудах больного направляют к кардиохирургу.
Синдром Марфана является редким генетическим заболеванием, которое сопровождается неправильным развитием соединительной ткани. Впоследствии у больного возникают поражения скелетных структур, сердца и сосудов, органов зрения, нервной и других систем организма. Постоянное врачебное наблюдение за такими пациентами, регулярное диагностическое обследование и своевременное лечение возникающих патологий позволяют существенно улучшать качество жизни таких людей и предупреждают развитие жизнеугрожающих осложнений.
Медицинская анимация о симптомах синдрома Марфана (англ. яз.):
О синдроме Марфана в программе «Жить здорово!» с Еленой Малышевой (см. с 30:20 мин.):
 (голосов - 1, среднее: 5,00 из 5)
(голосов - 1, среднее: 5,00 из 5)