Синдромом Барттера называют редко встречающуюся генетическую патологию мочевыделительной системы, при которой развивается выраженное нарушение щелочного равновесия, электролитного обмена, гиповолемия и компенсаторная гиперплазия почечного околоклубочкового аппарата. Это состояние провоцируется присутствием дефекта в петле Генле, наследуемом по аутосомно-рецессивному типу. Из-за такого изменения клетки почек не могут задерживать калий, и он постоянно утрачивается организмом, так как выводится с мочой. Синдром Барттера обычно дает о себе знать уже в детском возрасте и при прогрессировании может приводить к существенному ухудшению состояния больного, развитию нефрокальциноза и почечной недостаточности.
Причины и классификация
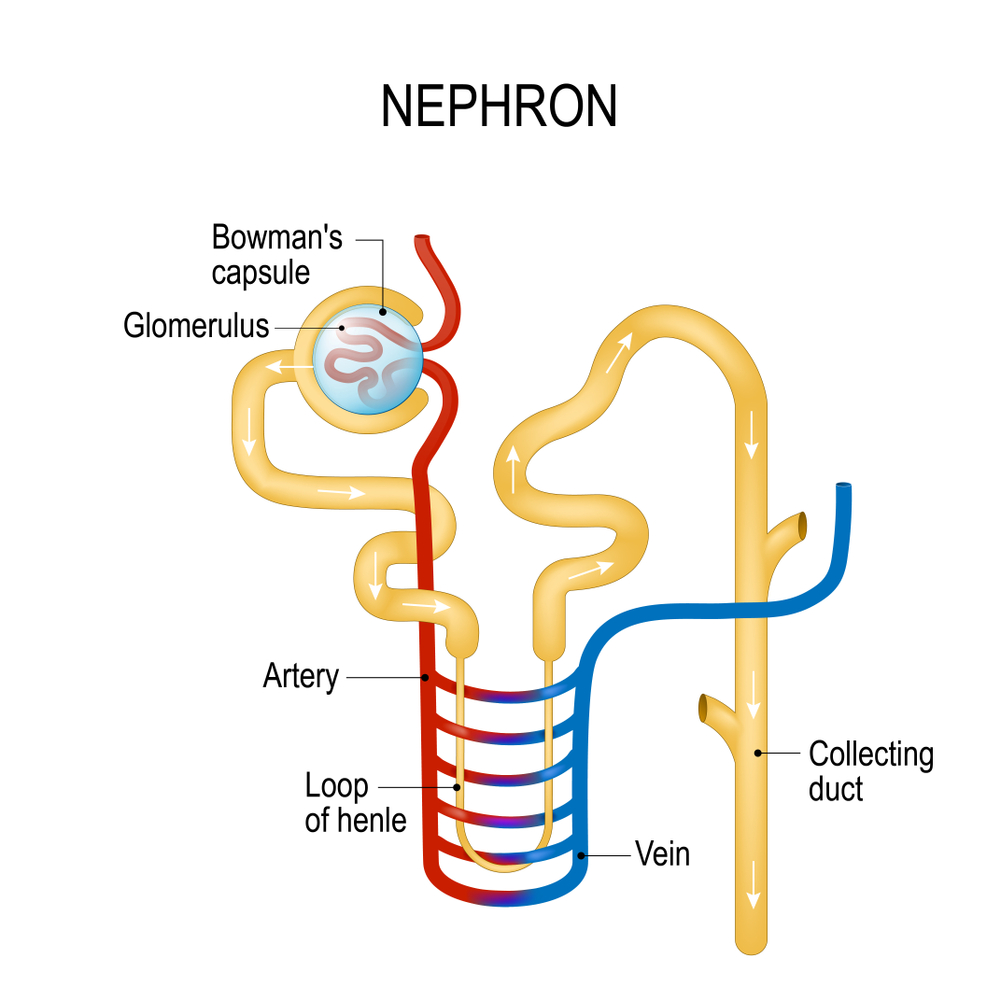
Главной причиной развития рассматриваемого синдрома является мутация генов, которая провоцирует изменение транспортной функции почечных канальцев. В зависимости от вида мутированных генов различают следующие разновидности этого недуга:
- неонатальный (или тип I) – вызывается мутацией NKCC2(15q) и дает о себе знать сразу после рождения, проявляется нефролитиазом кальциевого типа, тяжелой полиурией и гипертермией;
- тип II – вызывается мутацией ROMK(llq24), является классическим вариантом, проявляется судорогами, слабостью мышц и парестезиями;
- тип III – вызывается мутацией CLKNKB(1p36), приводит к полиурии, почечной недостаточности, но артериальная гипертензия не развивается;
- синдром Гительмана – вызывается мутацией NCCT(16ql3), нередко обнаруживается у взрослых, проявляется хроническими артралгиями, кальцификацией суставов, хрящей, радужки и склеры глаза, гипомагниемией, иногда может приводить к терминальной почечной недостаточности.
Кроме этих четырех вариантов может развиваться и синдром псевдо-Барттера. Такое состояние не вызывается мутациями генов и может провоцироваться хроническим муковисцидозом, нерациональным и продолжительным приемом мочегонных или слабительных средств, периодически возникающей рвотой или длительной хлордефицитной диетой. Нередко этот псевдосиндром выявляется у девушек или женщин, которые пытаются бороться с лишним весом при помощи изнурительных диет и приема мочегонных средств.
Симптомы
Первые проявления синдрома обычно выявляются у новорожденных или после первого года жизни. Они возникают из-за постоянной нехватки калия, который быстро выводится из организма с мочой. Выделяют такие общие симптомы синдрома:
- полиурия – большой объем выделяемой мочи, приводящий к развитию обезвоживания;
- поражение всех мышечных тканей – вялость скелетных, гладких мышц и сердечной мышцы, частые судороги и псевдопараличи;
- парестезии, ригидность конечностей – вызываются нарушениями в функционировании нервной системы;
- задержка в умственном и физическом развитии.
Несмотря на эти симптомы показатели артериального давления сохраняются в норме (или даже являются пониженными).
При неонатальном варианте синдрома признаки этого нарушения возникают еще во время внутриутробного развития. Такие беременности протекают тяжело, сопровождаются многоводием и могут заканчиваться преждевременным родоразрешением. У новорожденных выявляются следующие проявления патологии:
- сонливость;
- ребенок плохо сосет грудь, отказывается от еды;
- медленная прибавка веса или его потеря;
- гипертермия;
- слабость мышц;
- нарушения зрения и слуха;
- отставание в психомоторном развитии.
При классическом течении этого синдрома первые признаки недуга возникают после первого года жизни ребенка. У больного обнаруживаются следующие проявления патологии:
- полиурия;
- задержка развития и роста;
- нарушения в пищеварении: запоры, рвота;
- предрасположенность к обезвоживанию;
- патологическая сильная жажда (полидипсия).
При синдроме Гительмана рассматриваемая патология дает знать о себе уже после наступления 6 лет. У ребенка наблюдается повышенная утомляемость, слабость мышц и эпизоды возвратной тетании (спазмов мускулатуры). Однако этот вариант синдрома Барттера протекает более благоприятно, чем другие типы.
Диагностика

Заподозрить присутствие синдрома Барттера у ребенка врач может по таким признакам, как полиурия и мышечная слабость. В дальнейшем диагностика опирается на проведение ряда лабораторных анализов, позволяющих выявить множество отклонений, характерных для такой патологии.
Результаты анализов крови и мочи обнаруживают следующие отклонения:
- пониженный уровень ионов калия, хлора, магния и натрия в крови;
- повышенное содержание ионов натрия, калия, хлора и магния в моче;
- гипрекальциурия;
- гиперфосфатемия;
- повышение в крови уровня ренина и альдостерона;
- экскреция с мочой простагландинов и калликреина.
При таких отклонениях в показателях анализов артериальное давление остается в пределах нормы и не повышается.
При неонатальном варианте синдрома уже в первые 7 дней жизни новорожденного обнаруживаются следующие признаки:
- низкий удельный вес мочи;
- повышенное количество ионов калия, магния, хлора и натрия;
- метаболический алкалоз с гипокалиемией;
- повышенная активность ренина и альдостерона в крови;
- повышенный уровень простагландинов в моче и крови.
При классическом варианте синдрома у больного обнаруживается ненарушенная способность концентрировать мочу и метаболический алкалоз с нормальным или повышенным содержанием кальция. При синдроме Гительмана у пациентов выявляется резкое снижение уровня магния в крови и пониженное количество кальция в моче.
Лабораторная диагностика проводится при условии отсутствии утраты калия и хлоридов через пищеварительный тракт и при обстоятельствах, когда пациент не принимает слабительные и мочегонные средства.
В большинстве случаев для выявления синдрома Барттера достаточно проведения лабораторных анализов. Только иногда диагностика дополняется выполнением биопсии почки, при которой обнаруживается характерная для данной патологии гиперплазия околоклубочкового аппарата.
Для исключения ошибок при постановке диагноза синдром Барттера дифференцируют со следующими состояниями:
- неправильный прием диуретиков;
- хроническая рвота;
- сопровождающиеся дефицитом магния состояния;
- хроническая надпочечниковая недостаточность;
- изолированный гиперальдостеронизм.
Лечение
Основная цель лечения синдрома Барттера направляется на восполнение дефицита таких элементов, как калий и натрий. Для этого пациентам назначается заместительная и медикаментозная терапия. Кроме этого, больным необходимо соблюдать диету, которая обеспечивает достаточное поступление натрия и калия с продуктами питания, и дополнительно принимать препараты калия.
Пациентам назначаются следующие препараты:
- ингибиторы синтеза простагландинов: аспирин, индометацин и другие нестероидные противовоспалительные средства;
- ингибиторы АПФ для снижения секреции альдостерона и ренина: каптоприл;
- препараты магния: при синдроме Гительмана.
При неонатальной разновидности патологии сразу после рождения новорожденным проводят срочную заместительную терапию, заключающуюся в инфузионном вливании солевых растворов натрия и калия. Для этого применяются растворы NaCl и KCl. Для предотвращения потери калия применяются калийсберегающие мочегонные средства: триамтерен, амилорид, спиронолактон.
При присутствии у ребенка признаков недоношенности назначение индометацина, имеющего ряд нежелательных побочных реакций, не проводится до 1-1,5 месяцев.
При синдроме песвдо-Барттера лечение в первую очередь направлено на устранение первопричины нарушений. Дополнительно может назначаться триамтерен и амилорид.
При терминальной почечной недостаточности пациенту может проводиться трансплантация почки.
Прогноз
Синдром Барттера является генетической патологией и не может излечиваться полностью. Однако при раннем выявлении и проведении адекватного своевременного лечения во время новорожденности или в раннем возрасте специалистам удается стабилизировать состояние ребенка и уменьшать тяжесть проявлений и последствий. Например, правильно и вовремя проведенная заместительная и медикаментозная терапия позволяют смягчать проявления задержки в физическом и психическом развитии.
Тяжелое течение синдрома приводит к нефрокальцинозу, который становится причиной развития хронической почечной недостаточности. При отсутствии адекватного лечения неонатального типа патологии тяжелые электролитные нарушения и обезвоживание организма ребенка приводят к летальному исходу.
К какому врачу обратиться
При обильном и частом мочеиспускании, мышечной слабости и судорогах следует обратиться к нефрологу. После проведения анализов крови и мочи врач сможет поставить правильный диагноз и назначит необходимое лечение. В редких случаях обследование дополняется выполнением биопсии почки.
Синдром Барттера является наследственным заболеванием, и его развитие вызывается различными генными мутациями, которые приводят к тяжелым нарушениям щелочного равновесия, электролитного обмена, гиповолемии и компенсаторной гиперплазии околоклубочкового аппарата почек. При несвоевременном выявлении и лечении патология может приводить к развитию хронической почечной недостаточности и смерти больного. Для коррекции проявлений такой наследственной патологии почек проводится заместительная и медикаментозная терапия.