Синдром Бругада – кардиальная патология, характеризующаяся эпизодами потери сознания (связанными с тяжелыми аритмиями) и внезапной смертью у лиц без органических изменений в сердце. Впервые он был описан в медицинской литературе в 1992 году кардиологами испанского происхождения братьями Brugada, которые наблюдали течение болезни у трехлетней девочки из Польши, внезапно погибшей, несмотря на проводимую противоаритмическую терапию и имплантацию кардиостимулятора. Они обратили внимание на связь электрокардиологических проявлений с синкопальными состояниями и высоким риском внезапного летального исхода.
Синдром встречается во всем мире, но точная распространенность его в популяции неизвестна. Считается, что он чаще выявляется у представителей европеоидного этнического типа. Причем более подвержены заболеванию лица мужского пола (8-9:1). В России данная патология выявляется нечасто, однако ученые связывают это не с низким уровнем заболеваемости, а с недостаточной осведомленностью и меньшей ориентацией врачей на клинику синдрома, а также с наличием его скрытых форм.
Почему возникает

Синдром Бругада мало изучен и относится к числу наследственных заболеваний. Он передается от родителей к детям по аутосомно-доминантному типу с неполной пенетрантностью (проявляется в полной мере не у всех носителей дефектного гена). В основе его лежит генетическая аномалия регуляции работы ионных каналов (натриевых, кальциевых) в кардиомиоцитах. У большинства таких больных происходит мутация гена SCN5A, ответственного за функционирование натриевых каналов. Следует отметить, что мутация этого гена также может приводить к формированию синдрома удлиненного QT, поэтому две эти патологии считаются родственными.
Предполагается, что возникновение синдрома может быть обусловлено и другими мутациями, не все из которых точно идентифицированы. Благодаря достижениям современной науки удалось выявить 6 основных генов, изменения в которых вызывают развитие патологического процесса. Кроме вышеназванного гена к ним относят:
- GPD1L 3 хромосомы (кодирует активность фермента, участвующего в работе натриевых каналов);
- CACNA1C 12 хромосомы (ответственный за функционирование отдельных составляющих высокопороговых кальциевых каналов);
- CACNB2 10 хромосомы (кодирует β₂-субъединицу этих каналов);
- SCN4B 11 хромосомы (регулирует синтез белковых молекул, необходимых для работы малых натриевых каналов);
- SCN1B 19 хромосомы (координирует активность натриевых каналов).
У 25 % больных генетика синдрома остается не ясной, а в 15 % случаев они не имеют характерного семейного анамнеза.
Механизм развития заболевания и возникновения аритмии связывают с уменьшением количества натриевых каналов в кардиомиоцитах и ранней их инактивацией, что приводит к изменению проницаемости мембран этих клеток для ионов натрия. В результате нарушается формирование трансмембранного потенциала и страдает функция возбудимости и сократимости. Также определенную роль в этих процессах играет активность вегетативной нервной системы и дисбаланс между α- и β-адренергической стимуляцией. Усиливать проявления синдрома способны лекарственные средства, снижающие активность ионных каналов в сердце.
Симптомы
Клиническое течение при синдроме Бругада может быть различным: от абсолютно бессимптомного до клинически манифестного (с потерей сознания и эпизодами аритмии). Выраженность клинических проявлений зависит от степени повреждения натриевых каналов. Однако во всех случаях риск летального исхода остается высоким.
Дебют синдрома наблюдается в любом возрасте. Он проявляет себя как у маленьких детей, так и у пожилых. Но чаще первые признаки заболевания появляются в возрасте 30-40 лет. Обычно сначала обнаруживаются изменения на электрокардиограмме:
- блокада правой ножки пучка Гиса особой формы (постоянная или преходящая; полная или неполная);
- подъем сегмента ST в правых грудных отведениях (V₁-V₃);
- увеличение длительности интервала PR;
- пароксизмы желудочковой тахикардии.
Эти признаки могут появляться все сразу или существовать в разных комбинациях:
- у одних больных они выявляются постоянно;
- у других – появляются периодически;
- у третьих – их можно обнаружить только после медикаментозной или другой стимуляции.
Клинически синдром проявляется синкопальными состояниями, которые часто происходят в вечернее или ночное время (особенно во второй половине ночи), когда человек отдыхает или спит. Потере сознания предшествует:
- резкая слабость и бледность;
- ощущение сердцебиений;
- дискомфорт за грудиной;
- головокружение;
- потемнение в глазах;
- звон в ушах;
- холодный липкий пот.
Часто такие обмороки бывают кратковременными и продолжаются несколько секунд. За такое время человек не успевает понять, что с ним произошло и не всегда обращается за медицинской помощью. Но возможно и более глубокое нарушение сознания с судорогами и непроизвольным мочеиспусканием, при котором существенно возрастает риск летального исхода. При этом на ЭКГ регистрируется желудочковая тахикардия или фибрилляция желудочков. Приступы потери сознания могут провоцироваться:
- приемом лекарств;
- употреблением алкоголя;
- лихорадкой.
У некоторых больных синдром остается незамеченным длительное время. Иногда единственным проявлением болезни становится внезапная смерть. Если таких пациентов удается реанимировать, то при детальном изучении анамнеза у части из них выявляется наличие повторных обмороков неясного происхождения, которым не придавалось особого значения.
При обследовании у лиц, страдающих синдромом Бругада, отсутствуют структурные изменения со стороны сердца.
Клинические варианты
Общепринятой классификации данной патологии не существует, но на основании особенностей течения можно выделить несколько вариантов синдрома:
- полный (типичная ЭКГ и клиническая картина);
- бессимптомный, с характерными изменениями на ЭКГ, без отягощенного анамнеза и синкопе;
- бессимптомный, с типичным семейным анамнезом и ЭКГ-признаками;
- бессимптомный, в семье с полной формой синдрома, при котором изменения на ЭКГ появляются только после нагрузочных тестов;
- клинически манифестный без постоянных ЭКГ-проявлений, которые обнаруживаются только после проведения фармакологических проб.
Диагностика
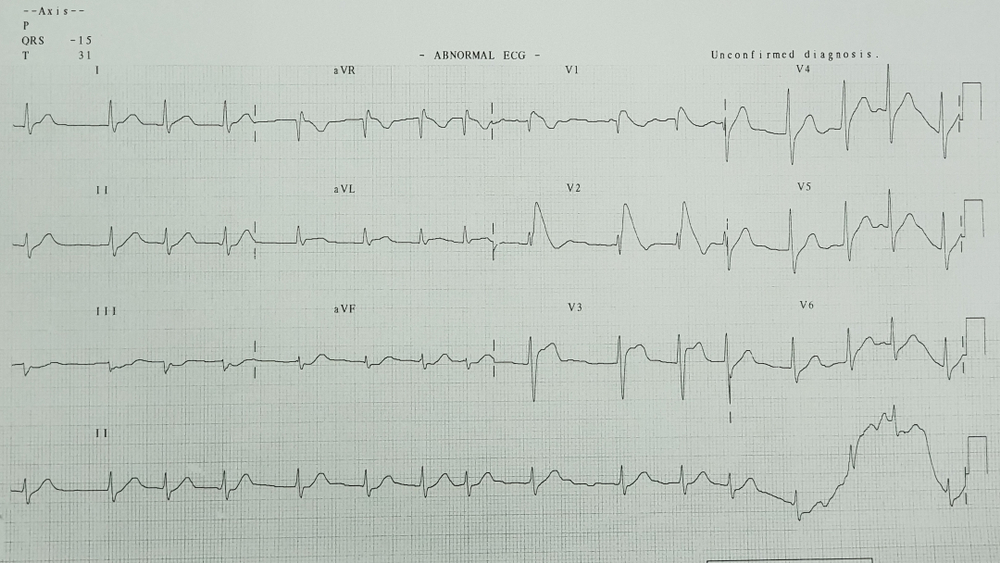
Диагностика синдрома сложна. Не всегда удается сразу сопоставить изменения на ЭКГ с клиническими и анамнестическими данными. Тем более у части пациентов для того, чтобы выявить ЭКГ-признаки, нужно провести исследование во время приступа, сразу после него или после стимуляции. Однако часто обращает на себя внимание наличие случаев внезапного летального исхода в семье, что служит поводом для обследования. Особенно важно не пропустить больных с эпизодами потери сознания или клинической смерти в анамнезе.
Основными методами диагностики при этом являются:
- электрокардиография;
- холтеровское мониторирование;
- молекулярно-генетические исследования;
- фармакологические пробы.
На последних остановимся подробнее. Они проводятся в условиях полной готовности к оказанию неотложной медицинской помощи, так как способствуют возникновению жизнеугрожающих аритмий. Для их выполнения пациенту вводят антиаритмики I А и C класса (прокаинамид, флекаинид, аймалин), после чего регистрируют электрокардиограмму.
В связи с наследственным характером заболевания необходимо тщательное обследование родственников больного для выявления скрытых и бессимптомных форм синдрома.
Дифференциальная диагностика проводится с целым рядом патологических состояний, имеющих сходные проявления на ЭКГ:
- аритмогенная дисплазия правого желудочка;
- синдром удлиненного QT;
- кардиомиопатии;
- расслаивающая аневризма аорты;
- передозировка некоторых лекарств (антидепрессантов);
- опухоль средостения и др.
Принципы лечения
Лечение при синдроме Бругада направлено на предупреждение приступов аритмии и внезапной смерти. Для этого применяется:
- медикаментозная терапия;
- имплантация кардиовертера-дефибриллятора.
Из медикаментов таким пациентам может назначаться:
- хинидин;
- амиодарон;
- изопротеренол;
- дизопирамид.
Эффективность такой терапии остается под вопросом. С помощью этих лекарственных средств не всегда удается добиться желаемых результатов. Другие противоаритмические средства при синдроме Бругада противопоказаны, особенно это касается препаратов I А и C класса.
Больным с симптомным вариантом синдрома Бругада показана имплантация кардиовертера-дефибриллятора. Только так у них можно предупредить летальный исход. Этот прибор способен самостоятельно анализировать сердечную деятельность и восстанавливать ритм, нанося электрический разряд, когда начинается фибрилляция желудочков. Также такой метод лечения показан больным:
- выжившим после реанимации;
- после пароксизма желудочковой аритмии;
- имеющим в семье случаи внезапной смерти.
Спорным остается вопрос об имплантации кардиостимулятора лицам с бессимптомным течением болезни.
К какому врачу обратиться
Пациенты с синдромом Бругада наблюдаются у кардиолога. Им назначается консультация кардиохирурга и генетика. Для исключения других причин обморочных состояний необходимо обследование у невролога.
Заключение
Прогноз при синдроме Бругада весьма серьезный. Больные часто умирают во время тяжелых приступов аритмии. Более 25 % случаев внезапной кардиальной смерти связаны именно с данной патологий. На современном этапе достоверно эффективным методом лечения, позволяющим повлиять на исход болезни, считается только имплантация кардиостимулятора. Но медицинская наука продолжает развиваться, и поиск новых методов лечения продолжается.
К. м. н. Т. А. Гончарик рассказывает о синдроме Бругада:
Наглядно о синдроме Бругада (англ. яз.):