Мукополисахаридозы объединяют группу наследственных заболеваний, в основе которых лежит нарушение обмена мукополисахаридов, обеспечивающих нормальное функционирование соединительной ткани. В медицинской литературе описано около 14 типов данной патологии.
Мукополисахаридоз 2 типа получил название синдром Хантера. Это заболевание, сцепленное с Х-хромосомой, имеющее рецессивный тип наследования. Как правило, выявляется синдром Хантера у мальчиков. В единичных случаях возможны случаи болезни у девочек, обусловленные инактивацией здоровой Х-хромосомы.
Причины
Непосредственной причиной данной патологии считается мутация в структурном гене лизосомного фермента (идуронат-2-сульфатазы), принимающего участие в синтезе мукополисахаридов. Современной медицине известно около 300 видов мутаций этого гена, большинство из них уникальны. Это могут быть мелкие или крупные делеции, перестройки гена и др. Такие мутации приводят к изменению строения или нарушению функциональной способности фермента и накоплению в лизосомах отдельных классов мукополисахаридов. Причем происходит это повсеместно:
- в хрящах;
- сухожилиях;
- надкостнице;
- сосудах;
- нервной ткани;
- печени и селезенке.
Особенности течения
Клинические проявления мукополисахаридоза 2 типа многообразны. При этом выраженность патологических симптомов может различаться. В клинической практике условно выделяют две формы болезни – легкую и тяжелую, хотя ее фенотипов существует намного больше.
Тяжелая форма синдрома дебютирует в возрасте 1-3 лет. Ее течение напоминает мукополисахаридоз 1 типа, но болезнь прогрессирует медленнее и ее симптомы менее выражены. У таких пациентов выявляют:
- изменение строения лицевого отдела черепа и туловища по типу гаргоилизма;
- отставание в росте;
- множественный костный дизостоз;
- тугоподвижность суставов;
- умственную отсталость;
- огрубение кожи и ее утолщение;
- избыточный рост волос;
- наличие гладких, лишенных волосяного покрова и напоминающих морскую гальку участков кожи в межлопаточной области, на боковых поверхностях груди, шеи;
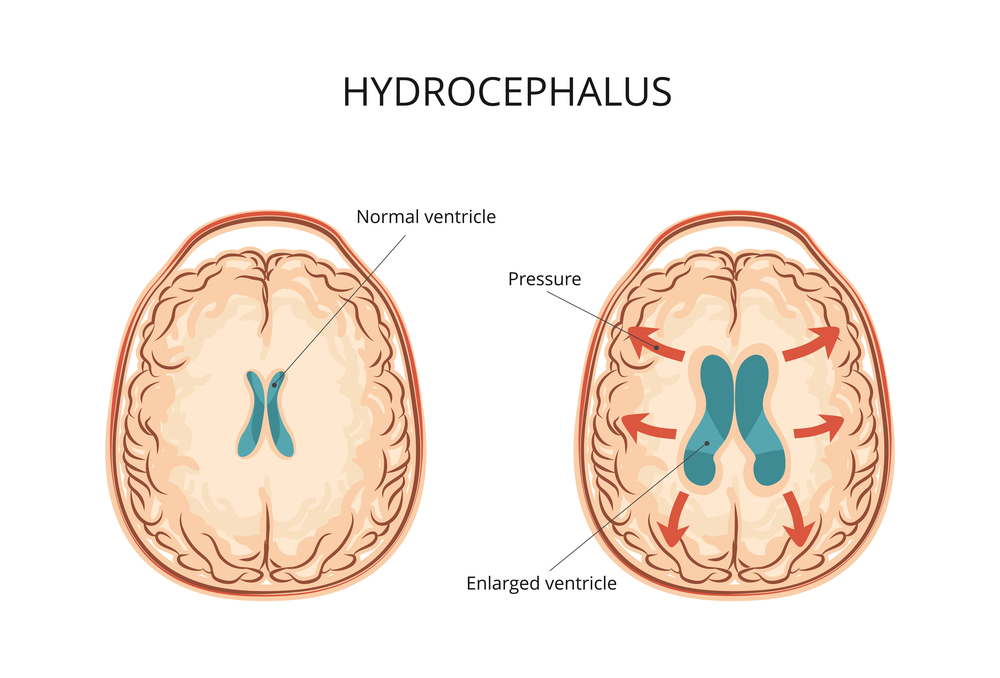
- признаки гидроцефалии;
- тугоухость;
- хроническую диарею;
- сердечную патологию и др.
Следует отметить, что при мукополисахаридозе 2 типа помутнение роговицы не встречается.
Термин «гаргоилизм» характеризует внешний вид больных мукополисахаридозом. Он произошел от названия фигур, изображенных на Соборе Парижской Богоматери во Франции. Типичными его проявлениями являются:
- грубые черты лица;
- нависающий лоб;
- толстые губы и язык;
- запавшая переносица;
- широкий нос с большими ноздрями;
- гипертелоризм глаз (широко расставленные глаза);
- низко расположенные ушные раковины;
- редкие зубы;
- небольшой рост;
- диспропорциональное строение скелета (короткое туловище и шея);
- костные деформации (черепа, грудины, позвоночника);
- большой живот и др.
Легкая форма синдрома Хантера проявляет себя несколько позже – в возрасте 3-8 лет, а иногда 10-15 лет. Такие дети имеют нормальный интеллект. Основными признаками заболевания являются:
- обструкция верхних отделов респираторного тракта;
- сердечные пороки;
- ограничение подвижности в суставах;
- нарушение слуха.
Если при тяжелой форме мукополисахаридоза летальный исход наступает на втором десятилетии жизни в результате неврологических осложнений, то при его легкой форме продолжительность жизни варьирует в широких пределах и зависит от тяжести поражения внутренних органов. Наиболее частая причина смерти таких больных – недостаточность кровообращения.
Принципы диагностики и лечения

Заподозрить синдром Хантера врач может, детально изучив жалобы больного, историю его заболевания и оценив его внешний вид.
Подтверждает диагноз биохимическое исследование с определением уровня гликозаминогликанов, выделяемых с мочой. Если этот уровень оказывается высоким, то на следующем этапе оценивается активность идуронат-2-сульфатазы, содержащейся в лейкоцитах или кожных фибробластах.
В сложных случаях может проводиться ДНК-анализ. Однако эта процедура является слишком непростой и продолжительной, поэтому применяется редко.
В семьях с высоким риском возникновения данной патологии рекомендуется проведение пренатальной диагностики с определением активности лизосомного фермента в амниотической жидкости или ворсинах хориона на ранних стадиях развития плода.
Дифференциальная диагностика при синдроме Хантера проводится с другими лизосомными болезнями накопления.
Тактика ведения больных зависит от тяжести течения синдрома и поражения внутренних органов. В большинстве случаев лечение симптоматическое. При тяжелых формах болезни таким пациентам может назначаться идурсульфаза. Применение этого препарата ограничено в связи с высокой стоимостью и необходимостью частого введения (еженедельно).
К какому врачу обратиться
За детьми с синдромом Хантера осуществляется наблюдение в крупных специализированных медицинских центрах; по месту жительства они лечатся под контролем педиатра. Дополнительно назначаются консультации кардиолога, невролога, гастроэнтеролога, специалиста по слухопротезированию, логопеда, специалиста по лечебной физкультуре.
Заключение
Синдром Хантера – тяжелое заболевание, снижающее качество жизни больных и часто являющееся причиной летального исхода. Средняя продолжительность жизни таких пациентов составляет около 30 лет. Эффективных и доступных методов лечения этого заболевания в настоящее время не существует. Поэтому особое внимание уделяется вопросам профилактики и пренатальной диагностики.
Видеоруководство по синдрому Хантера:
 (Пока оценок нет)
(Пока оценок нет)